Progenesis QI Proteomics and ProteinLynx Global Server giving different IDs from same MSe data - WKB29713
SYMPTOMS
- When the same data is processed with the same processing parameters in PLGS3.0.3 and Progenesis QI Proteomics v3 or v4, the protein identification results are different
- All processing parameters (for example, Apex3D low energy and high energy thresholds, lock mass) are confirmed to be identical
- All search parameters are identical (same fasta file, digest reagent, peptide and fragment mass tolerance, allowed modifications, allowed missed cleavages)
ENVIRONMENT
- PLGS3.0.3
- Progenesis QI for Proteomics v3 or v4
NOTE : If comparing PLGS3.0.3 and Progenesis QI for Proteomics v4, make sure that the peptide3D modification described in article 13412 has been applied to PLGS3.0.3. This modification is implemented by default in Progenesis QI for Proteomics v4 and can have an impact on the proteins identified.
CAUSE
Difference in the retention time window processed
When setting up an MSe experiment in Progenesis QIp, there are two places were the retention time range must be specified. For MSe ID searches, you must define the retention time window in the MSe data import wizard. For the Progenesis difference engine, you define the retention time window in the Automatic Processing wizard. If you only define the second of these, the retention time window defined in the Automatic Processing wizard is not applied to the MSe peak processing used to identify proteins. This means that all the data is processed by Apex3D and peptide3D and used in the iadbs search, so the search in Progenesis QIp has not been performed with the same subset of data used in PLGS. However, the RT limits in the Automatic processing are correctly applied to the data used for quantification.
FIX or WORKAROUND
Reprocess the data in Progenesis QIp using the same RT window that you used in the PLGS processing parameters.
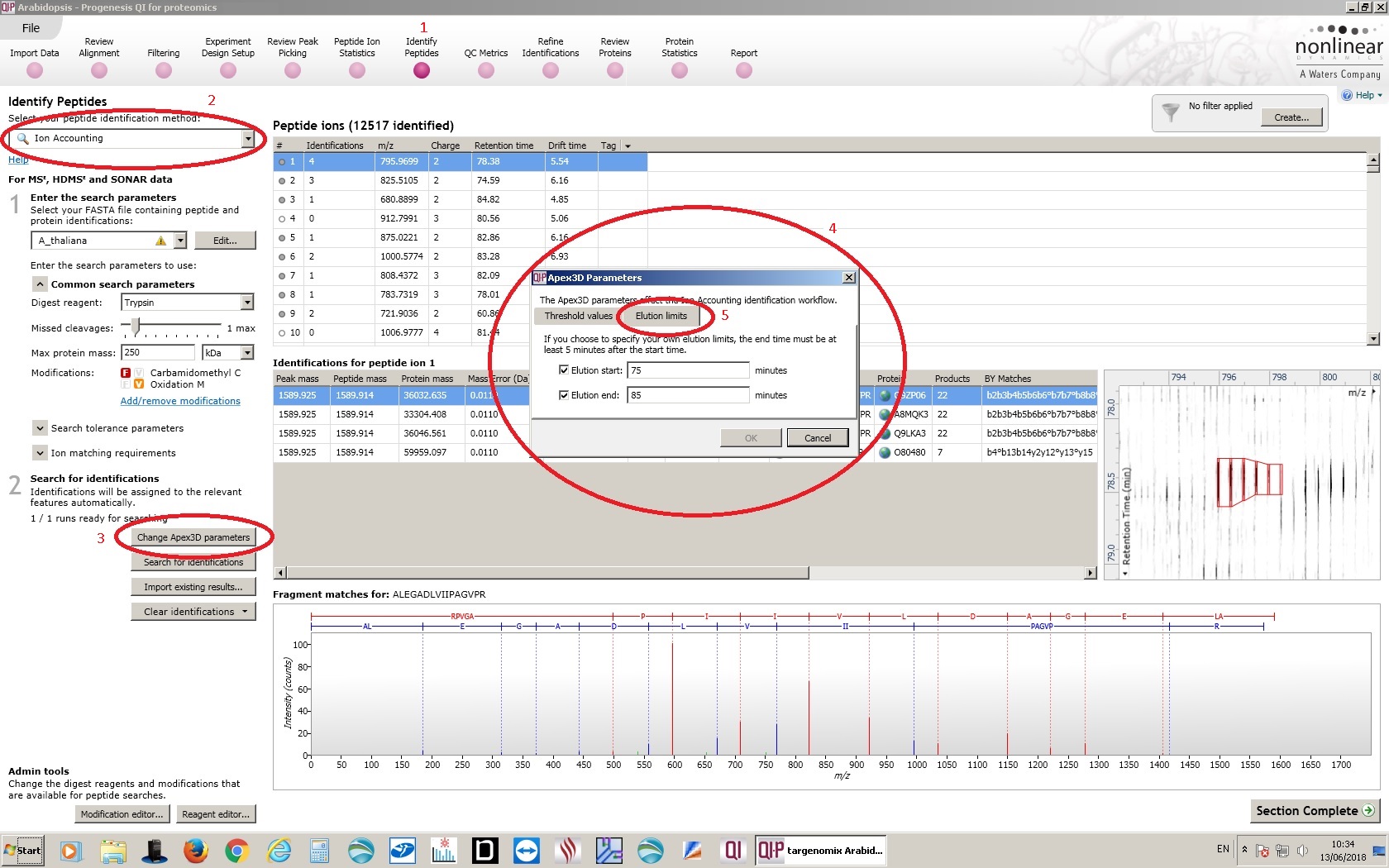
- Navigate to the Identify proteins tab in Progenesis QI for Proteomics.
- Make sure that Ion Accounting is the selected peptide identification method.
- Click the Change Apex3D Parameters button.
- This opens the Apex3D parameters wizard, which is roughly equivalent to the Processing parameters in PLGS. Make sure that the Threshold values are the same as those used on PLGS.
- Select the Elution limits tab, and set the same retention time window that you set in the processing parameters in PLGS.
- Click OK to reprocess the data. NOTE: You will see no progress bar while the data is being reprocessed. You can monitor the Apex3D and peptide3D processes in the Windows task manager. This may take a long time.
- When Apex3D and Peptide3D have completed for all samples in the analysis, click the "Search for Identifications" button to repeat the search step (equivalent of PLGS workflow) against the data from the define retention time window.
ADDITIONAL INFORMATION
To ensure that you are comparing like with like, you must use the correct versions of PLGS and Progenesis QI for Proteomics; otherwise, the versions of the processing executable used will be different. In terms of processing executables:
Progenesis QIp v4 = PLGS3.0.3 with the peptide3D modification described in WKB13412
Progenesis QIp v3 = Unmodified PLGS3.0.3
Progenesis QIp v3 with "rolled back" MSe processing exes = PLGS3.0.2
Progenesis QIp v2 = PLGS 3.0.2